
| Hội chứng Kearns-Sayre | |
|---|---|
| Tên khác | oculocraniosomatic disorder, oculocraniosomatic neuromuscular disorder with ragged red fibers |
 | |
| Kearns–Sayre syndrome is inherited in an autosomal recessive manner(or mitochondrial inheritence) | |
| Khoa/Ngành |
Nhãn khoa |
Hội chứng Kearns–Sayre (KSS) là một bệnh cơ ty lạp thể với khởi phát điển hình trước 20 tuổi. KSS là một biến thể hội chứng nghiêm trọng hơn của nhãn khoa ngoài tiến triển mạn tính (viết tắt CPEO), một hội chứng được đặc trưng bởi sự tham gia cô lập của các cơ kiểm soát chuyển động của mí mắt (levator palpebrae, orbicularis ocular). Điều này dẫn đến ptosis và ophthalmoplegia tương ứng. KSS liên quan đến sự kết hợp của CPEO đã được mô tả cũng như bệnh võng mạc sắc tố ở cả hai mắt và bất thường dẫn truyền tim. Các triệu chứng khác có thể bao gồm atebia ataxia, yếu cơ gần, điếc, đái tháo đường, thiếu hormone tăng trưởng, suy tuyến cận giáp và bệnh lý nội tiết. Trong cả hai bệnh này, sự tham gia của cơ bắp có thể bắt đầu đơn phương nhưng luôn phát triển thành thâm hụt song phương, và quá trình diễn ra tiến triển. Thảo luận này được giới hạn cụ thể cho các biến thể nghiêm trọng hơn và có liên quan hệ thống.
Dấu hiệu và triệu chứng
Các cá nhân mắc KSS ban đầu có biểu hiện tương tự như những người có CPEO điển hình. Khởi phát là trong thập kỷ thứ nhất và thứ hai của cuộc đời.
Triệu chứng đầu tiên của bệnh này là đơn phương ptosis, hoặc khó mở mí mắt, dần dần tiến triển thành ptosis hai bên. Khi ptosis xấu đi, cá nhân thường mở rộng cổ, nâng cằm trong nỗ lực ngăn mí mắt che khuất trục thị giác. Cùng với sự phát triển ngấm ngầm của ptosis, chuyển động của mắt cuối cùng bị hạn chế khiến một người phụ thuộc nhiều hơn vào việc xoay đầu sang bên hoặc lên xuống để xem các vật thể trong ngoại vi trường thị giác.
Bệnh võng mạc sắc tố

KSS dẫn đến một sắc tố của võng mạc, chủ yếu ở phía sau fundus. Sự xuất hiện được mô tả như là một vẻ ngoài "muối và hạt tiêu". Có sự phân tán lan tỏa của biểu mô sắc tố võng mạc với tác dụng lớn nhất xảy ra tại macula. Điều này trái ngược với viêm võng mạc sắc tố nơi sắc tố là ngoại vi. Sự xuất hiện của võng mạc trong KSS tương tự như đã thấy trong loạn trương lực cơ loại 1 (viết tắt DM1). Chứng mù đêm khiêm tốn có thể gặp ở bệnh nhân mắc KSS. Mất thị lực thường nhẹ và chỉ xảy ra ở 40-50% bệnh nhân.
Bất thường dẫn truyền tim
Những điều này thường xảy ra nhiều năm sau khi phát triển ptosis và nhãn khoa.Block nhĩ thất là thâm hụt dẫn truyền tim phổ biến nhất. Điều này thường tiến triển thành block tim độ ba, đó là sự tắc nghẽn hoàn toàn của sự dẫn điện từ tâm nhĩ đến tâm thất. Các triệu chứng của khối tim bao gồm ngất, không dung nạp vận động và nhịp tim chậm.
Thiếu folate não
Bệnh nhân Kearns-Sayre luôn bị phát hiện thiếu folate não, một hội chứng trong đó mức 5-MTHF bị giảm trong dịch não tủy mặc dù bình thường trong huyết thanh. Điều trị bằng folinic acid trong một số trường hợp có thể làm giảm bớt các triệu chứng liên quan và điều chỉnh một phần bất thường não liên quan, đặc biệt là nếu bắt đầu sớm trong quá trình bệnh. Nguyên nhân được đề xuất của tình trạng thiếu folate não trong hội chứng Kearns-Sayre là do các cơ chế trong màng đệm chịu trách nhiệm truyền folate từ huyết thanh vào dịch não tủy.
Khác
Như đặc trưng trong ấn phẩm gốc của Kearns năm 1965 và trong các ấn phẩm sau này, các đặc điểm không phù hợp của KSS có thể xảy ra là yếu cơ mặt, hầu họng, cơ bắp và tứ chi, giảm thính lực, tầm vóc nhỏ, thay đổi điện não đồ, tiểu não ataxia và mức độ cao của dịch não tủy.
Nguyên nhân
Hội chứng Kearns- Sayre xảy ra tự phát trong phần lớn các trường hợp. Trong một số trường hợp, nó đã được chứng minh là di truyền qua ty thể, trội hoàn toàn tự phát hoặc di truyền lặn tự phát. Không có thiên hướng về chủng tộc hay giới tính, và không có yếu tố rủi ro nào được biết đến. Tính đến năm 1992, chỉ có 226 trường hợp được báo cáo trong tài liệu xuất bản.
Di truyền
KSS là kết quả của việc xóa trong DNA ty thể (mtDNA) gây ra một sinh thái di truyền cụ thể của dấu hiệu y tế và các triệu chứng. mtDNA được truyền độc quyền từ noãn của mẹ. DNA ti thể bao gồm 37 gen được tìm thấy trong vòng tròn nhiễm sắc thể có chiều dài 16,569 cặp cơ sở. Trong số này, 13 gen mã hóa protein của chuỗi vận chuyển điện tử (viết tắt là "ETC"), 22 mã hóa RNA truyền (tRNA) và hai gen mã hóa các tiểu đơn vị lớn và nhỏ tạo thành RNA ribosome (rRNA). 13 protein liên quan đến ETC của ty thể là cần thiết cho phosphoryl hóa oxy hóa. Đột biến trong các protein này dẫn đến việc sản xuất năng lượng bị suy yếu bởi ty thể. Sự thiếu hụt năng lượng tế bào này biểu hiện dễ dàng nhất ở các mô phụ thuộc nhiều vào quá trình trao đổi chất hiếu khí như não, cơ xương và cơ tim, cơ quan cảm giác và thận. Đây là một yếu tố liên quan đến việc trình bày các bệnh về ty thể.
Có những yếu tố khác liên quan đến biểu hiện của một bệnh ty thể bên cạnh kích thước và vị trí của một đột biến. Ty thể sao chép trong mỗi lần phân chia tế bào trong thời kỳ mang thai và trong suốt cuộc đời. Bởi vì đột biến trong bệnh ty thể thường xảy ra sớm trong thời kỳ mang thai trong những bệnh này, nên chỉ những ty thể trong dòng dõi bị đột biến là khiếm khuyết. Điều này dẫn đến sự phân bố không đồng đều của ty thể rối loạn chức năng trong mỗi tế bào và giữa các mô khác nhau của cơ thể. Điều này mô tả thuật ngữ dị thể đặc trưng của các bệnh về ty thể bao gồm KSS. Sự phân bố mtDNA đột biến trong mỗi tế bào, mô và cơ quan, phụ thuộc vào thời điểm và nơi xảy ra đột biến. Điều này có thể giải thích tại sao hai bệnh nhân có đột biến giống hệt nhau trong mtDNA có thể xuất hiện với các kiểu hình hoàn toàn khác nhau và lần lượt các hội chứng khác nhau. Một ấn phẩm vào năm 1992 bởi Fischel-Ghodsian đã xác định xóa 4.977 bp giống nhau trong mtDNA ở hai bệnh nhân có hai bệnh hoàn toàn khác nhau. Một trong những bệnh nhân có KSS đặc trưng, trong khi bệnh nhân còn lại mắc một bệnh rất khác gọi là Hội chứng tụy –tủy xương Pearson. Phức tạp hóa vấn đề, trong một số trường hợp, hội chứng Pearson đã được chứng minh là tiến triển thành KSS sau này trong cuộc sống.
Nhiều nghiên cứu gần đây đã kết luận rằng sự nhân đôi mtDNA cũng có thể đóng một vai trò quan trọng trong việc xác định kiểu hình nào hiện diện. Bản sao của mtDNA dường như là đặc trưng của tất cả các trường hợp mắc hội chứng KSS và Pearson, trong khi chúng không có trong CPEO.
Việc xóa mtDNA trong KSS có kích thước khác nhau (1,3-8kb), cũng như vị trí trong bộ gen ty thể. Việc xóa phổ biến nhất là 4,9kb và kéo dài từ vị trí 8469 đến vị trí 13147 trên bộ gen. Việc xóa này hiện diện ở khoảng người bị KSS
Chẩn đoán
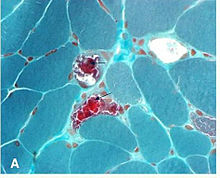
Một bác sĩ thần kinh thị giác thường tham gia vào chẩn đoán và quản lý KSS. Một cá nhân nên bị nghi ngờ có KSS dựa trên kết quả khám lâm sàng. Sự nghi ngờ đối với bệnh cơ nên được tăng lên ở những bệnh nhân bị nhãn khoa không khớp với một nhóm bệnh thần kinh sọ cụ thể (liệt dây thần kinh mắt, liệt dây thần kinh thứ tư, liệt dây thần kinh thứ tư. Ban đầu, các nghiên cứu hình ảnh thường được thực hiện để loại trừ các bệnh lý phổ biến hơn. Chẩn đoán có thể được xác nhận bằng sinh thiết cơ và có thể được bổ sung xác định PCR các đột biến mtDNA.
Kết quả sinh thiết
Không cần thiết phải sinh thiết một cơ mắt để chứng minh các bất thường mô bệnh học. Mặt cắt ngang của các sợi cơ được nhuộm bằng Gmöri trichrom stain được xem bằng kính hiển vi quang học. Trong các sợi cơ chứa tỷ lệ cao của ty thể bị đột biến, có nồng độ ty thể cao hơn. Điều này làm cho các sợi này có màu đỏ đậm hơn, làm cho hình dạng tổng thể của sinh thiết được mô tả là "sợi đỏ bị rách. Sự bất thường cũng có thể được chứng minh trong các mẫu sinh thiết cơ bằng các nghiên cứu mô hóa học khác như nhuộm enzyme ty thể kính hiển vi, phân tích sinh hóa của mô cơ (tức là hoạt động của enzyme chuỗi vận chuyển điện tử) và bằng cách phân tích DNA ty thể của cơ. "
Nghiên cứu trong phòng thí nghiệm
Nồng độ lactate và pyruvate thường tăng lên do tăng chuyển hóa yếm khí và giảm tỷ lệ ATP: ADP. Phân tích CSF cho thấy mức protein tăng cao, thường là >100 mg/dl, cũng như mức độ lactate.
Quản lý
Hiện tại không có điều trị chữa bệnh cho KSS. Bởi vì đây là một tình trạng hiếm gặp, chỉ có báo cáo trường hợp điều trị với rất ít dữ liệu để hỗ trợ hiệu quả của chúng. Một số khám phá đầy hứa hẹn đã được báo cáo có thể hỗ trợ cho việc khám phá các phương pháp điều trị mới với nghiên cứu sâu hơn. Các tế bào vệ tinh chịu trách nhiệm tái tạo sợi cơ. Nó đã được lưu ý rằng mtDNA đột biến là hiếm hoặc không thể phát hiện trong các tế bào vệ tinh được nuôi cấy từ bệnh nhân mắc KSS. Shoubridge và cộng sự. (1997) đã đặt câu hỏi liệu mtDNA wildtype có thể được phục hồi vào mô cơ hay không bằng cách khuyến khích tái tạo cơ bắp. Trong nghiên cứu đã đề cập, các sợi cơ tái tạo được lấy mẫu tại vị trí sinh thiết ban đầu, và người ta thấy rằng về cơ bản chúng là homoplasmic cho mtDNA wildtype. Có lẽ với các kỹ thuật trong tương lai để thúc đẩy tái tạo tế bào cơ và tăng sinh tế bào vệ tinh, tình trạng chức năng ở bệnh nhân KSS có thể được cải thiện đáng kể.
Một nghiên cứu đã mô tả một bệnh nhân mắc KSS đã giảm nồng độ coenzyme Q10 trong huyết thanh. Quản lý 60–120 mg Coenzyme Q10 trong 3 tháng dẫn đến bình thường hóa lactate và pyruvate, cải thiện chẩn đoán trước đây khối AV độ thứ nhất và cải thiện chuyển động mắt.
Một ECG sàng lọc được khuyến nghị ở tất cả các bệnh nhân trình bày với CPEO. Trong KSS, cấy ghép máy tạo nhịp được khuyên dùng sau khi phát triển bệnh dẫn truyền quan trọng, ngay cả ở những bệnh nhân không có triệu chứng.
Nên kiểm tra các rối loạn nội tiết, bao gồm đo nồng độ glucose huyết thanh, xét nghiệm chức năng tuyến giáp, nồng độ calci và magnesi và nồng độ điện giải trong huyết thanh. Tăng aldosteron được thấy ở 3% bệnh nhân KSS.
Lịch sử
Bộ ba CPEO, bệnh võng mạc sắc tố hai bên và bất thường dẫn truyền tim được mô tả lần đầu tiên trong một báo cáo trường hợp của hai bệnh nhân vào năm 1958 bởi Thomas P. Kearns (1922-2011), MD., and George Pomeroy Sayre (1911-1992), MD. Một trường hợp thứ hai đã được xuất bản vào năm 1960 bởi Jager và các đồng tác giả báo cáo các triệu chứng này ở một cậu bé 13 tuổi. Các trường hợp bệnh nhân CPEO trước đây đột ngột tử vong đã được công bố, đôi khi được ghi nhận là do rối loạn nhịp tim. Các trường hợp khác đã ghi nhận một sắc tố đặc biệt của võng mạc, nhưng không có ấn phẩm nào trong số các ấn phẩm này ghi nhận ba bệnh lý này xảy ra cùng nhau như một hội chứng di truyền. Kearns đã xuất bản một trường hợp xác định vào năm 1965 mô tả 9 trường hợp không liên quan với bộ ba này. Năm 1988, kết nối đầu tiên được thực hiện giữa KSS và xóa quy mô lớn DNA ty thể cơ (viết tắt mtDNA) Kể từ khám phá này, nhiều lần xóa DNA ti thể đã được liên kết với sự phát triển của KSS.
Tham khảo
Liên kết ngoài
- kearns_sayre tại Viện Rối loạn Thần kinh và Đột quỵ Quốc gia Hoa Kỳ (NINDS)
- Kearns Sayre syndrome tại trụ sở của Viện Sức khỏe Quốc gia Hoa Kỳ (NIH) về Bệnh hiếm gặp
Bản mẫu:Bệnh ti thể Bản mẫu:Bệnh học mắt Bản mẫu:Bệnh về mối nối cơ và cơ