
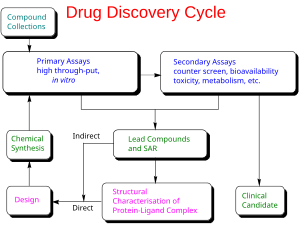
Phát triển thuốc là quá trình đưa một loại dược phẩm mới ra thị trường một khi hợp chất đầu đã được xác định thông qua quá trình phát hiện thuốc. Nó bao gồm các nghiên cứu tiền lâm sàng trên các vi sinh vật và động vật, nộp đơn xin tình trạng pháp lý, chẳng hạn như thông qua Hoa Kỳ Food and Drug Administration cho một loại thuốc mới nghiên để bắt đầu thử nghiệm lâm sàng trên con người, và có thể bao gồm các bước có được sự chấp thuận quy định với một ứng dụng thuốc mới để tiếp thị thuốc.
Phát triển thực thể hóa học mới
Nhìn rộng ra, quá trình phát triển thuốc có thể được chia thành công việc lâm sàng và tiền lâm sàng.

Tiền lâm sàng
Các thực thể hóa học mới (NCE, còn được gọi là các thực thể phân tử mới hoặc NMEs) là các hợp chất xuất hiện từ quá trình khám phá thuốc. Chúng có hoạt động hứa hẹn chống lại một mục tiêu sinh học cụ thể quan trọng trong bệnh tật. Tuy nhiên, rất ít thông tin về sự an toàn, độc tính, dược động học và chuyển hóa của NCE này ở người. Đây là chức năng của sự phát triển thuốc để đánh giá tất cả các thông số này trước khi thử nghiệm lâm sàng ở người. Mục tiêu chính khác của sự phát triển thuốc là khuyến nghị liều lượng và lịch trình cho lần sử dụng đầu tiên trong thử nghiệm lâm sàng ở người ("người đầu tiên " [FIM] hoặc Liều người đầu tiên [FHD]).
Ngoài ra, phát triển thuốc phải thiết lập các tính chất hóa lý của NCE: thành phần hóa học, tính ổn định và độ hòa tan của nó. Các nhà sản xuất phải tối ưu hóa quy trình họ sử dụng để tạo ra hóa chất để họ có thể mở rộng quy mô từ một nhà hóa học dược phẩm sản xuất vài miligam, đến sản xuất trên quy mô kilôgam và tấn. Họ tiếp tục kiểm tra các sản phẩm cho phù hợp để đóng gói như viên nang, viên nén, bình phun, tiêm bắp, tiêm dưới da hoặc tiêm tĩnh mạch công thức. Cùng với nhau, các quá trình này được biết đến trong sự phát triển lâm sàng và tiền lâm sàng là chemistry, manufacturing, and control (CMC).
Nhiều khía cạnh của phát triển thuốc tập trung vào việc đáp ứng các yêu cầu quy định của cơ quan cấp phép thuốc. Chúng thường tạo thành một số thử nghiệm được thiết kế để xác định độc tính chính của một hợp chất mới trước khi sử dụng lần đầu tiên ở người. Yêu cầu pháp lý là phải đánh giá độc tính cơ quan chính (ảnh hưởng đến tim và phổi, não, thận, gan và hệ tiêu hóa), cũng như tác động lên các bộ phận khác của cơ thể có thể bị ảnh hưởng bởi thuốc (ví dụ, da nếu thuốc mới được chuyển qua da). Càng ngày, các xét nghiệm này được thực hiện bằng phương pháp in vitro (ví dụ, với các tế bào biệt lập), nhưng nhiều xét nghiệm chỉ có thể được thực hiện bằng cách sử dụng động vật thí nghiệm để chứng minh sự tương tác phức tạp của quá trình trao đổi chất và phơi nhiễm thuốc với độc tính.
Thông tin được thu thập từ thử nghiệm tiền lâm sàng này, cũng như thông tin về CMC, và nộp cho cơ quan quản lý (ở Mỹ, FDA), dưới dạng đơn đăng ký Điều tra về Thuốc Mới (IND). Nếu IND được phê duyệt, sự phát triển sẽ chuyển sang giai đoạn lâm sàng.
Giai đoạn lâm sàng
Thử nghiệm lâm sàng bao gồm ba hoặc bốn bước:
- Thử nghiệm giai đoạn I, thường ở những người tình nguyện khỏe mạnh, xác định sự an toàn và liều lượng.
- Các thử nghiệm ở giai đoạn II được sử dụng để có được kết quả ban đầu về hiệu quả và khám phá thêm về sự an toàn ở một số lượng nhỏ bệnh nhân mắc bệnh do NCE nhắm tới.
- Các thử nghiệm pha III là các thử nghiệm lớn, quan trọng để xác định tính an toàn và hiệu quả ở một số lượng lớn bệnh nhân mắc bệnh mục tiêu. Nếu an toàn và hiệu quả được chứng minh đầy đủ, thử nghiệm lâm sàng có thể dừng ở bước này và NCE tiến tới giai đoạn ứng dụng thuốc mới (NDA).
- Các thử nghiệm giai đoạn IV là các thử nghiệm sau phê duyệt đôi khi là một điều kiện gắn liền với FDA, còn được gọi là nghiên cứu giám sát sau thị trường.
Quá trình xác định các đặc tính của thuốc không dừng lại khi NCE bắt đầu thử nghiệm lâm sàng ở người. Ngoài các xét nghiệm cần thiết để lần đầu tiên đưa một loại thuốc mới vào phòng khám, các nhà sản xuất phải đảm bảo rằng mọi độc tính lâu dài hoặc mãn tính đều được xác định rõ, bao gồm các tác động lên các hệ thống không được theo dõi trước đó (khả năng sinh sản, sinh sản, hệ thống miễn dịch, trong số những người khác). Họ cũng phải kiểm tra hợp chất về khả năng gây ung thư (xét nghiệm gây ung thư).
Nếu một hợp chất xuất hiện từ các thử nghiệm này với hồ sơ an toàn và độc tính có thể chấp nhận được và công ty có thể cho thấy nó có hiệu quả mong muốn trong các thử nghiệm lâm sàng, thì danh mục bằng chứng NCE có thể được đệ trình để phê duyệt tiếp thị ở các quốc gia khác nhau nơi nhà sản xuất lên kế hoạch để bán nó Tại Hoa Kỳ, quy trình này được gọi là "ứng dụng thuốc mới" hoặc NDA.
Hầu hết các NCE thất bại trong quá trình phát triển thuốc, vì chúng có độc tính không thể chấp nhận được hoặc vì đơn giản là chúng không có tác dụng đối với bệnh mục tiêu như trong các thử nghiệm lâm sàng.
Xu hướng thu thập dấu ấn sinh học và thông tin di truyền từ những người tham gia thử nghiệm lâm sàng và tăng đầu tư của các công ty trong lĩnh vực này, dẫn đến năm 2018 chiếm một nửa trong số tất cả các thử nghiệm thuốc thu thập thông tin này, tỷ lệ hiện mắc trên 80% trong các thử nghiệm ung thư.
Giá cả
Toàn bộ chi phí đưa một loại thuốc mới (tức là thực thể hóa học mới) ra thị trường - từ khám phá qua thử nghiệm lâm sàng đến phê duyệt - rất phức tạp và gây tranh cãi. Thông thường, các công ty chi hàng chục đến hàng trăm triệu đô la Mỹ. Một yếu tố của sự phức tạp là những con số cuối cùng được công bố rộng rãi thường không chỉ bao gồm chi phí tự trả để thực hiện một loạt các thử nghiệm lâm sàng giai đoạn I-III, mà còn cả chi phí vốn trong thời gian dài (10 năm trở lên) trong thời gian đó công ty phải trang trải chi phí tự trả cho việc khám phá thuốc tiền lâm sàng. Ngoài ra, các công ty thường không báo cáo liệu một con số cụ thể có bao gồm chi phí vốn hóa hay chỉ bao gồm chi phí tự trả hoặc cả hai. Một yếu tố phức tạp khác là tất cả các ước tính đều dựa trên việc phát hành tự nguyện các thông tin bí mật khác có thể không dễ dàng được xác minh độc lập.
Một nghiên cứu năm 2010 đã đánh giá cả chi phí vốn hóa và chi phí tự trả khi đưa một loại thuốc mới ra thị trường lần lượt là khoảng 1,8 tỷ đô la Mỹ và 870 triệu đô la Mỹ.
Trong một phân tích về chi phí phát triển thuốc cho 98 công ty trong một thập kỷ, chi phí trung bình cho mỗi loại thuốc được phát triển và phê duyệt bởi một công ty thuốc đơn lẻ là 350 triệu đô la. Nhưng đối với các công ty đã phê duyệt từ 8 đến 13 loại thuốc trong hơn 10 năm, chi phí cho mỗi loại thuốc lên tới 5,5 tỷ USD, chủ yếu do mở rộng địa lý để tiếp thị và chi phí liên tục cho các thử nghiệm Giai đoạn IV và theo dõi liên tục để đảm bảo an toàn.
Các lựa chọn thay thế cho phát triển thuốc thông thường có mục tiêu cho các trường đại học, chính phủ và ngành công nghiệp dược phẩm hợp tác và tối ưu hóa các nguồn lực.
Đánh giá
Bản chất của một dự án phát triển thuốc được đặc trưng bởi tỷ lệ tiêu hao cao, chi tiêu vốn lớn và thời gian dài. Điều này làm cho việc định giá các dự án và công ty như vậy là một nhiệm vụ đầy thách thức. Không phải tất cả các phương pháp định giá có thể đối phó với các đặc thù này. Các phương pháp định giá được sử dụng phổ biến nhất là giá trị hiện tại ròng được điều chỉnh theo rủi ro (rNPV), cây quyết định, tùy chọn thực hoặc so sánh.
Các trình điều khiển giá trị quan trọng nhất là chi phí vốn hoặc tỷ lệ chiết khấu được sử dụng, các thuộc tính pha như thời lượng, tỷ lệ thành công và chi phí và doanh thu được dự báo, bao gồm chi phí hàng hóa và chi phí tiếp thị và bán hàng. Các khía cạnh khách quan ít hơn như chất lượng quản lý hoặc tính mới của công nghệ nên được phản ánh trong ước tính dòng tiền.