
| Một phần của một loạt bài về |
| Đại dịch COVID-19 |
|---|
 |
|
|
|
Các vấn đề
Các vấn đề và hạn chế
Ảnh hưởng kinh tế – xã hội
|
|
|
Phát triển thuốc COVID-19 là quá trình nghiên cứu để phát triển vắc-xin phòng ngừa hoặc thuốc theo toa điều trị có khả năng làm giảm mức độ nghiêm trọng của bệnh coronavirus 2019 (COVID-19). Trên phạm vi quốc tế tính đến tháng 3 năm 2020, khoảng 100 công ty dược phẩm, công ty công nghệ sinh học, nhóm nghiên cứu đại học và tổ chức y tế đã tham gia vào các giai đoạn phát triển vắc-xin hoặc thuốc này.Tổ chức Y tế Thế giới (WHO),Cơ quan Dược phẩm Châu Âu (EMA),Cục Quản lý Thực phẩm và Dược phẩm Hoa Kỳ (FDA), và chính phủ Trung Quốc và các nhà sản xuất thuốc đang phối hợp với học thuật và các nhà nghiên cứu trong ngành để tăng tốc độ phát triển vắc-xin, thuốc kháng vi-rút và liệu pháp kháng thể đơn dòng.
Đến tháng 3 năm 2020, Liên minh đổi mới phòng chống dịch bệnh (CEPI) đã khởi xướng một quỹ phát triển vắc-xin COVID-19 quốc tế, với mục tiêu huy động 2 tỷ đô la Mỹ cho nghiên cứu và phát triển vắc-xin, và cam kết đầu tư 100 triệu đô la Mỹ vào vắc-xin phát triển trên nhiều quốc gia. Đầu tháng 3, chính phủ Canada đã công bố tài trợ 275 triệu đô la Canada cho 96 dự án nghiên cứu về các biện pháp đối phó y tế chống lại COVID-19, bao gồm nhiều ứng cử viên vắc-xin tại các trường đại học Canada, với kế hoạch thành lập một "ngân hàng vắc-xin" quốc gia chứa các vắc-xin mới để đưa ra thị trường nếu một đợt bùng phát coronavirus khác xảy ra. Nhiều hợp chất chống vi-rút được thiết lập để điều trị các bệnh nhiễm trùng khác đang được tái sử dụng hoặc trong các nỗ lực nghiên cứu lâm sàng mới để làm giảm bớt bệnh COVID-19, kể từ tháng 3 năm 2020.
Phát triển vắc-xin và thuốc là một quá trình gồm nhiều giai đoạn, thường cần hơn năm năm để đảm bảo an toàn và hiệu quả của hợp chất vắcxin/thuốc mới. Vào tháng 2 năm 2020, WHO cho biết họ không mong đợi vắc-xin chống lại SARS-CoV-2 – virus gây bệnh COVID-19 – sẽ xuất hiện trong vòng chưa đầy 18 tháng, và ước tính thời gian cần thiết để chứng minh sự an toàn và hiệu quả của vắc-xin là một năm. Một số cơ quan quản lý quốc gia, chẳng hạn như EMA và FDA, đã phê duyệt các thủ tục để tiến hành thử nghiệm lâm sàng.
Đến cuối tháng 3 năm 2020, ba liệu pháp chống virus tiềm năng – favipiravir, remdesivir và ritonavir – đã đến giai đoạn thử nghiệm cuối cùng ở người – Thử nghiệm lâm sàng giai đoạn III – và một số vắc-xin có thể đã bước vào giai đoạn đầu của đánh giá an toàn con người, giai đoạn I. Vào ngày 21 tháng 3, Trung tâm kiểm soát và phòng ngừa dịch bệnh Hoa Kỳ (CDC) đã đưa ra một lời khuyên của bác sĩ liên quan đến remdesivir cho những người nhập viện vì viêm phổi do COVID-19: "Trong khi các thử nghiệm lâm sàng rất quan trọng để thiết lập sự an toàn và hiệu quả của thuốc này, các bác sĩ lâm sàng không được tiếp cận thử nghiệm lâm sàng có thể yêu cầu remdesivir được sử dụng mở rộng thông qua nhà sản xuất cho bệnh nhân bị viêm phổi lâm sàng. "
Quá trình
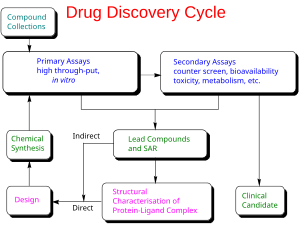
Phát triển thuốc là quá trình đưa vắc-xin bệnh truyền nhiễm hoặc thuốc điều trị mới ra thị trường một khi hợp chất chì đã được xác định thông qua quá trình phát hiện thuốc. Nó bao gồm nghiên cứu trong phòng thí nghiệm về vi sinh vật và động vật, nộp đơn xin tình trạng quy định, chẳng hạn như thông qua FDA, cho một loại thuốc mới điều tra để bắt đầu thử nghiệm lâm sàng trên người, và có thể bao gồm bước để có được sự chấp thuận theo quy định với một đăng ký thuốc mới để bán loại thuốc này. Toàn bộ quá trình – từ ý tưởng thông qua thử nghiệm tiền lâm sàng trong phòng thí nghiệm đến phát triển thử nghiệm lâm sàng, bao gồm cả thử nghiệm giai đoạn I-III – đến vắc-xin hoặc thuốc được phê duyệt thường mất hơn một thập kỷ.
Thực thể hóa học mới
Phát triển vắc-xin COVID-19 hoặc thuốc chống vi-rút điều trị bắt đầu bằng việc kết hợp một khái niệm hóa học với cơ chế dự phòng tiềm năng của vắc-xin hoặc hoạt động chống virus trong tương lai.

Thiết kế thuốc và thử nghiệm trong phòng thí nghiệm
Các thực thể hóa học mới (NCE, còn được gọi là các thực thể phân tử mới hoặc NMEs) là các hợp chất xuất hiện từ quá trình khám phá thuốc để chỉ định một ứng cử viên vắc-xin hoặc thuốc chống virus. Chúng có hoạt động đầy hứa hẹn chống lại một mục tiêu sinh học liên quan đến bệnh COVID-19. Khi bắt đầu phát triển vắc-xin hoặc thuốc, người ta biết rất ít về sự an toàn, độc tính, dược động học và chuyển hóa của NCE ở người. Đây là chức năng và nghĩa vụ của sự phát triển thuốc để đánh giá tất cả các thông số này trước khi thử nghiệm lâm sàng ở người để chứng minh tính an toàn và hiệu quả. Mục tiêu chính khác của sự phát triển thuốc là khuyến nghị liều lượng và lịch trình cho lần sử dụng đầu tiên trong thử nghiệm lâm sàng ở người ("first in human" [FIH] hoặc First human dose [FHD], trước đây còn được gọi là "first in man "[FIM]).
Ngoài ra, phát triển thuốc phải thiết lập các tính chất hóa lý của NCE: trang điểm hóa học, tính ổn định và độ hòa tan của nó. Các nhà sản xuất phải tối ưu hóa quy trình họ sử dụng để tạo ra hóa chất để họ có thể mở rộng quy mô từ một nhà hóa học dược phẩm sản xuất vài miligam, đến sản xuất trên quy mô kilôgam và tấn. Họ tiếp tục kiểm tra các sản phẩm cho phù hợp với gói như viên nang, viên nén, bình phun, tiêm bắp, tiêm dưới da hoặc tiêm tĩnh mạch. Cùng với nhau, các quá trình này được biết đến trong sự phát triển lâm sàng và tiền lâm sàng là hóa học, sản xuất và kiểm soát (chemistry, manufacturing, and control - CMC).
Nhiều khía cạnh của phát triển thuốc tập trung vào việc đáp ứng các yêu cầu quy định của cơ quan cấp phép thuốc. Chúng thường tạo thành các xét nghiệm được thiết kế để xác định độc tính chính của một hợp chất mới trước khi sử dụng lần đầu tiên ở người. Yêu cầu quy định là phải đánh giá độc tính cơ quan chính (ảnh hưởng đến tim và phổi, não, thận, gan và hệ tiêu hóa), cũng như tác động lên các bộ phận khác của cơ thể có thể bị ảnh hưởng bởi thuốc (ví dụ, da nếu được tiêm vắc-xin mới bằng cách tiêm da). Càng ngày, các xét nghiệm này càng được thực hiện bằng phương pháp in vitro (ví dụ, với các tế bào biệt lập), nhưng nhiều xét nghiệm chỉ có thể được thực hiện bằng cách sử dụng động vật thí nghiệm để chứng minh sự tương tác phức tạp của quá trình trao đổi chất và phơi nhiễm thuốc với độc tính.
Thông tin được thu thập từ thử nghiệm tiền lâm sàng này, cũng như thông tin về CMC, và nộp cho các cơ quan quản lý (ở Mỹ, FDA), dưới dạng Đơn xin cấp phép thuốc mới (IND) hoặc Đơn xin cấp phép Sinh học cho một loại vắc-xin. Nếu IND được phê duyệt, sự phát triển sẽ chuyển sang giai đoạn lâm sàng, và tiến trình thực hiện ở người – nếu một loại vắc-xin đang được phát triển ở Hoa Kỳ – được FDA theo dõi trong một "quy trình phê duyệt vắc-xin".
Các giai đoạn thử nghiệm lâm sàng
Các chương trình thử nghiệm lâm sàng bao gồm ba giai đoạn kéo dài nhiều năm đối với phê duyệt sản phẩm và giai đoạn thứ tư, sau phê duyệt để theo dõi an toàn vắc-xin hoặc điều trị bằng thuốc:
- Thử nghiệm pha I, thường ở những người tình nguyện khỏe mạnh, xác định sự an toàn và liều lượng.
- Các thử nghiệm ở giai đoạn II được sử dụng để thiết lập kết quả ban đầu về hiệu quả và khám phá thêm về sự an toàn ở một số ít người mắc bệnh do NCE nhắm đến.
- Các thử nghiệm pha III là các thử nghiệm lớn, quan trọng để xác định tính an toàn và hiệu quả ở số lượng người đủ lớn bị nhiễm COVID-19. Nếu an toàn và hiệu quả được chứng minh đầy đủ, thử nghiệm lâm sàng có thể dừng ở bước này và NCE tiến tới giai đoạn ứng dụng thuốc mới (NDA) để bắt đầu tiếp thị.
- Các thử nghiệm giai đoạn IV là các thử nghiệm sau phê duyệt có thể là một điều kiện gắn liền với FDA, còn được gọi là nghiên cứu giám sát sau khi đưa ra thị trường. Cho đến khi vắc-xin được cung cấp cho toàn bộ dân cư, tất cả các sự kiện bất lợi tiềm ẩn vẫn chưa được xác định, đòi hỏi vắc-xin phải trải qua các nghiên cứu pha IV với các báo cáo thường xuyên của Nhà sản xuất cho Hệ thống báo cáo về tác dụng phụ của vắc-xin (VAERS) để xác định các vấn đề sau khi bắt đầu sử dụng.
Quá trình xác định các đặc tính của thuốc không dừng lại khi NCE bắt đầu thử nghiệm lâm sàng ở người. Ngoài các xét nghiệm cần thiết để lần đầu tiên đưa vắc-xin hoặc thuốc chống vi-rút mới vào phòng khám, các nhà sản xuất phải đảm bảo rằng mọi độc tính lâu dài hoặc mãn tính đều được xác định rõ, bao gồm cả các tác dụng trên các hệ thống không được theo dõi trước đó (khả năng sinh sản, hệ thống miễn dịch, và các hệ thống khác).
Nếu một ứng cử viên vắc-xin hoặc hợp chất chống vi-rút qua được các xét nghiệm này với hồ sơ an toàn và độc tính chấp nhận được, và nhà sản xuất có thể cho thấy nó có hiệu quả mong muốn trong các thử nghiệm lâm sàng, thì danh mục bằng chứng NCE có thể được đệ trình để phê duyệt tiếp thị ở các quốc gia khác nhau nơi nhà sản xuất có kế hoạch bán nó. Tại Hoa Kỳ, quy trình này được gọi là "ứng dụng thuốc mới" hoặc NDA.
Tỷ lệ thất bại
Hầu hết các NCE thất bại trong quá trình phát triển thuốc, vì chúng có độc tính không thể chấp nhận được hoặc vì đơn giản là chúng không chứng minh được hiệu quả đối với căn bệnh mục tiêu, như thể hiện trong các thử nghiệm lâm sàng giai đoạn II-III. Một đánh giá năm 2018 chỉ ra rằng các thử nghiệm lâm sàng thất bại do kinh phí không đủ, điểm yếu trong thiết kế thử nghiệm và thực hiện thử nghiệm kém.
Một nghiên cứu bao gồm nghiên cứu lâm sàng trong những năm 1980-90 cho thấy chỉ có 21,5% ứng cử viên thuốc bắt đầu thử nghiệm giai đoạn I cuối cùng đã được chấp thuận cho phép đưa ra thị trường. Trong giai đoạn 2006-15, tỷ lệ thành công trong việc đạt được phê duyệt từ Giai đoạn I đến các thử nghiệm Giai đoạn III thành công trung bình dưới 10% và đặc biệt là 11,5% đối với vắc-xin ("sinh học"). Tỷ lệ thất bại cao liên quan đến phát triển dược phẩm được gọi là "tỷ lệ tiêu hao", đòi hỏi các quyết định trong giai đoạn đầu phát triển thuốc để "tiêu diệt" các dự án sớm để tránh các thất bại tốn kém.
Chi phí
Một nghiên cứu năm 2010 đã đánh giá cả chi phí vốn hóa và chi phí tự trả khi đưa một loại thuốc mới ra thị trường lần lượt là khoảng 1,8 tỷ đô la Mỹ và 870 triệu đô la Mỹ. Ước tính chi phí trung bình của các thử nghiệm 2015-16 để phát triển 10 loại thuốc chống ung thư là 648 triệu đô la Mỹ. Trong năm 2017, chi phí trung bình của một thử nghiệm quan trọng trên tất cả các chỉ định lâm sàng là 19 triệu đô la Mỹ.
Chi phí trung bình (tính trên đô la Mỹ năm 2013) của mỗi giai đoạn nghiên cứu lâm sàng là 25 triệu đô la Mỹ cho nghiên cứu an toàn giai đoạn I, 59 triệu đô la cho nghiên cứu hiệu quả ngẫu nhiên có kiểm soát giai đoạn II và 255 triệu đô la Mỹ cho thử nghiệm giai đoạn III để chứng minh tính tương đương hoặc ưu việt của nó đến một loại thuốc đã được phê duyệt hiện có, có thể lên tới 345 triệu đô la Mỹ. Chi phí trung bình để thực hiện thử nghiệm giai đoạn III 2015-16 cho một ứng cử viên thuốc điều trị bệnh truyền nhiễm là 22 triệu đô la Mỹ.
Toàn bộ chi phí đưa một loại thuốc mới (tức là thực thể hóa học mới) ra thị trường - từ khám phá qua thử nghiệm lâm sàng đến phê duyệt - rất phức tạp và gây tranh cãi. Trong một đánh giá năm 2016 của 106 ứng cử viên thuốc được đánh giá qua các thử nghiệm lâm sàng, tổng chi phí vốn cho một nhà sản xuất thuốc được phê duyệt qua các thử nghiệm giai đoạn III thành công là 2,6 tỷ đô la (năm 2013 đô la), số tiền tăng với tỷ lệ hàng năm là 8,5%. Trong giai đoạn 2003-2013 đối với các công ty đã phê duyệt 8-13 loại thuốc, chi phí cho mỗi loại thuốc có thể tăng lên tới 5,5 tỷ USD, chủ yếu do mở rộng địa lý quốc tế để tiếp thị và chi phí liên tục cho các thử nghiệm giai đoạn IV để giám sát an toàn liên tục.
Các lựa chọn thay thế cho phát triển thuốc thông thường có mục tiêu cho các trường đại học, chính phủ và ngành công nghiệp dược phẩm hợp tác và tối ưu hóa các nguồn lực.
Đồng bộ hóa việc phát triển thuốc COVID-19
Trong năm 2018,20, các sáng kiến mới để kích thích phát triển vắc-xin và thuốc kháng vi-rút bao gồm sự hợp tác giữa các tổ chức chính phủ và ngành công nghiệp, như Sáng kiến Thuốc sáng tạo Châu Âu,"Sáng kiến Con đường Quan trọng" của Hoa Kỳ để tăng cường đổi mới phát triển thuốc, và chỉ định Liệu pháp Đột phá để thúc đẩy sự phát triển và đánh giá theo quy định đối với các loại thuốc ứng cử viên đầy triển vọng. Đến tháng 3 năm 2020, Liên minh Quốc tế về Đổi mới Chuẩn bị Dịch tễ học (CEPI) cam kết đầu tư nghiên cứu 100 triệu đô la Mỹ trên một số quốc gia, và đưa ra lời kêu gọi khẩn cấp để tăng và đầu tư nhanh chóng 2 tỷ đô la để phát triển vắc-xin. Đầu năm 2020, nhiều hợp chất chống vi-rút được thành lập để điều trị các bệnh nhiễm trùng khác đã được tái sử dụng hoặc phát triển trong các nỗ lực nghiên cứu lâm sàng mới để làm giảm bớt bệnh COVID-19.
Vắc-xin đã được sản xuất chống lại một số bệnh do coronavirus gây ra cho động vật, bao gồm cả vi-rút viêm phế quản truyền nhiễm ở chim, coronavirus chó và coronavirus mèo.
Những nỗ lực trước đây để phát triển vắc-xin cho vi-rút trong họ coronaviridae có ảnh hưởng đến con người đã nhắm đến hội chứng hô hấp cấp tính nặng coronavirus (SARS) và hội chứng hô hấp Trung Đông (MERS). Vắc-xin chống SARS và MERS đã được thử nghiệm trên các mô hình động vật trong phòng thí nghiệm. Kể từ năm 2020, không có thuốc chữa bệnh hoặc vắc-xin bảo vệ cho SARS được chứng minh là an toàn và hiệu quả ở người. Xác định và phát triển các loại vắc-xin và thuốc mới để điều trị SARS là ưu tiên hàng đầu của chính phủ và các cơ quan y tế công cộng trên toàn thế giới.
Cũng không có vắc-xin đã được chứng minh chống lại MERS. Khi MERS trở nên phổ biến, người ta tin rằng nghiên cứu SARS hiện tại có thể cung cấp một khuôn mẫu hữu ích để phát triển vắc-xin và phương pháp điều trị chống lại nhiễm trùng MERS-CoV. Tính đến tháng 3 năm 2020, có một loại vắc-xin MERS (dựa trên DNA) đã hoàn thành thử nghiệm lâm sàng giai đoạn I ở người, và ba loại khác đang được tiến hành, tất cả đều là vắc-xin có vec-tơ, hai loại vec-tơ adenovirus (ChAdOx1-MERS, BVRS-GamVac) và một vectơ MVA (MVA-MERS-S).
Ứng cử viên cho vắc-xin và thuốc kháng virus
SARS-CoV-2 được xác định vào cuối năm 2019 là nguyên nhân của cái mà sau này được đặt tên là COVID-19. Một ổ dịch lớn lan rộng khắp thế giới vào năm 2020, dẫn đến hoạt động đầu tư và nghiên cứu đáng kể để phát triển vắc-xin. Nhiều tổ chức đang sử dụng bộ gen được công bố để phát triển các loại vắc-xin có thể chống lại SARS-CoV-2. Tổng cộng có khoảng 100 công ty và tổ chức học thuật (tháng 3 năm 2020), và khoảng 300 nghiên cứu tiền lâm sàng hoặc lâm sàng đang được tiến hành, kể từ tháng 3 năm 2020, bao gồm việc tái sử dụng nhiều hợp chất chống vi-rút được phê duyệt để điều trị các bệnh nhiễm trùng khác.
Nghiên cứu lâm sàng thất bại
Ở những người trưởng thành bị nhiễm COVID-19 nhập viện nặng ở Vũ Hán, Trung Quốc, điều trị bằng cách sử dụng kết hợp thuốc kháng virus – lopinavir - ritonavir (liệu pháp HIV/AIDS) – không mang lại lợi ích gì.
Ứng viên vắc-xin
Kể từ tháng 3 năm 2020, hai thử nghiệm lâm sàng an toàn giai đoạn I trên các ứng cử viên vắc-xin đã được khởi động:
- mRNA-1273: Viện Dị ứng và Bệnh truyền nhiễm Quốc gia Hoa Kỳ (NIAID) đã hợp tác với Moderna để phát triển một loại vắc-xin RNA phù hợp với sự tăng đột biến của bề mặt coronavirus. Vào tháng 2 năm 2020, NIAID đã đăng ký một nghiên cứu pha I về ứng cử viên vắc-xin Moderna có tên mRNA-1273 sẽ được tiến hành tại Seattle, Washington. Vào ngày 16 tháng 3 năm 2020, nghiên cứu của con người bắt đầu.
- Ad5-nCoV: * Một thử nghiệm an toàn giai đoạn I của một ứng cử viên vắc-xin adenovirus tái tổ hợp được sản xuất bởi CanSino Biologics Inc. (Thiên Tân, Trung Quốc), được gọi là Ad5-nCoV, đã bắt đầu tuyển dụng 108 người trưởng thành khỏe mạnh ở Vũ Hán, Trung Quốc vào tháng 3, với nghiên cứu kéo dài đến cuối năm 2020.
Một số ứng cử viên vắc-xin khác đã được theo dõi nhanh để bắt đầu nghiên cứu đầu tiên ở người, kể từ tháng 3 năm 2020. Vào cuối tháng 3 năm 2020, hàng chục công ty, nhóm nghiên cứu học thuật và liên minh quốc tế đang trong giai đoạn đầu thiết kế vắc-xin, thử nghiệm trong phòng thí nghiệm hoặc lập kế hoạch nghiên cứu an toàn giai đoạn I để tiến tới một ứng cử viên vắc-xin, với việc chính phủ Canada đầu tư 192 triệu đô la Canada vào việc nghiên cứu và phát triển vắc-xin.
Phòng chống phơi nhiễm
Dựa trên kinh nghiệm với thuốc chống vi trùng, điều trị dự phòng phơi nhiễm trước và điều trị dự phòng sau phơi nhiễm (PEP) với thuốc kháng vi-rút có thể là thủ tục hiệu quả để giảm thiểu nhiễm trùng bởi COVID-19. PEP sử dụng kháng sinh, rifampicin, được WHO khuyến cáo cho những người có nguy cơ nhiễm trùng cao trước hoặc sau khi tiếp xúc với đại dịch cúm. Thuốc kháng vi-rút được sử dụng ngay sau khi xuất hiện triệu chứng nhiễm COVID-19 có thể làm giảm bệnh tật và giảm nguy cơ lây nhiễm cho người khác bằng cách giảm sự phát tán của virus trong dịch tiết đường hô hấp.
Thuốc chống siêu vi: Giai đoạn III
- Favipiravir: Ở những người mắc COVID-19, favipiravir (được bán dưới dạng Avigan và được chấp thuận sử dụng ở Nhật Bản vào năm 2014 đối với một số bệnh do virus) đã được tìm thấy là an toàn và có hiệu quả sơ bộ trong thử nghiệm giai đoạn đầu ở Thâm Quyến, Trung Quốc.
- Remdesivir: ứng cử viên thuốc chống vi-rút của Gilead Science đang trong giai đoạn thử nghiệm lâm sàng, với kết quả sơ bộ dự kiến vào tháng 5 năm 2020, và đã được đưa ra trong hai thử nghiệm hiệu quả giai đoạn III tại Hồng Kông, Singapore, Hàn Quốc và Hoa Kỳ Tháng 3 năm 2020. Vào ngày 21 tháng 3, Trung tâm kiểm soát và phòng ngừa dịch bệnh Hoa Kỳ (CDC) đã đưa ra một lời khuyên của bác sĩ liên quan đến remdesivir cho những người bị viêm phổi do COVID-19: "Trong khi các thử nghiệm lâm sàng rất quan trọng để thiết lập sự an toàn và hiệu quả của thuốc này, các bác sĩ lâm sàng không có truy cập vào một thử nghiệm lâm sàng có thể yêu cầu remdesivir được sử dụng mở rộng thông qua nhà sản xuất cho bệnh nhân bị viêm phổi lâm sàng."
- ASC-09 + ritonavir (viên uống): ASC-09 (sản phẩm của công ty Trung Quốc, Ascletis Pharma Inc.), một chất ức chế protease HIV-1 kết hợp với ritonavir, đã được bắt đầu trong thử nghiệm giai đoạn III tại Trung Quốc trong tháng 2 năm 2020 nhằm đánh giá hiệu quả chống lại COVID-19.
Các hợp chất khác
- Chloroquine hoặc Hydroxychloroquine: Vào cuối tháng 1 năm 2020 trong đại dịch coronavirus 2019-20, các nhà nghiên cứu y học Trung Quốc khẳng định rằng nghiên cứu thăm dò chloroquine và hai loại thuốc khác, remdesivir và lopinavir/ritonavir, dường như có "tác dụng ức chế khá tốt" trên coronavirus mới 2019. Chloroquine cũng đã được đề xuất như là một phương pháp điều trị SARS-CoV với xét nghiệm in vitro ức chế virus.
Vào ngày 19 tháng 2 năm 2020, kết quả sơ bộ cho thấy chloroquine có thể hiệu quả và an toàn trong điều trị viêm phổi liên quan đến COVID-19. Sở Khoa học và Công nghệ Quảng Đông và Ủy ban Y tế và sức khỏe tỉnh Quảng Đông đã đưa ra một báo cáo nói rằng chloroquine phosphate "cải thiện tỷ lệ thành công của điều trị và rút ngắn thời gian nằm viện của bệnh nhân" và đề nghị cho những người được chẩn đoán viêm phổi coronavirus mới nhẹ, trung bình và nghiêm trọng sử dụng thuốc này.
Chloroquine đã được các cơ quan y tế Trung Quốc, Hàn Quốc và Ý khuyên dùng để điều trị COVID-19, mặc dù các cơ quan này và CDC Hoa Kỳ lưu ý cần chống chỉ định cho những người mắc bệnh tim hoặc tiểu đường. Vào tháng 2 năm 2020, cả hai loại thuốc đã được chứng minh là có hiệu quả làm giảm bệnh COVID-19, nhưng một nghiên cứu tiếp theo đã kết luận rằng hydroxychloroquine mạnh hơn chloroquine và có hồ sơ an toàn tốt hơn. Kết quả sơ bộ từ một thử nghiệm chỉ ra rằng chloroquine có hiệu quả và an toàn trong viêm phổi COVID-19, "cải thiện phát hiện hình ảnh phổi, thúc đẩy chuyển đổi âm tính với virus và rút ngắn quá trình điều trị bệnh." Hydroxychloroquine thường có sẵn hơn là chloroquine ở Hoa Kỳ.
Theo CDC Hoa Kỳ, nên sử dụng chloroquine hoặc hydroxychloroquine để điều trị cho những người nhập viện bị nhiễm COVID-19 ở một số quốc gia, mặc dù tại Hoa Kỳ không có khuyến nghị nào như vậy, kể từ tháng 3 năm 2020. Các thử nghiệm lâm sàng sơ bộ để đánh giá sự an toàn và hiệu quả của hydroxychloroquine trong điều trị nhiễm COVID-19 đã được lên kế hoạch tại Hoa Kỳ, nhưng CDC tuyên bố rằng "việc sử dụng, dùng thuốc hoặc thời gian điều trị bằng hydroxychloroquine để điều trị dự phòng hoặc điều trị nhiễm SARS-CoV-2" là không phù hợp tại thời điểm này.
- BDB-1: một kháng thể đơn dòng chống C5a (sản phẩm của Staidson Pharmaceuticals, Bắc Kinh) đang trong giai đoạn thử nghiệm lâm sàng giai đoạn II ở Trung Quốc.
- Brilacidin: một sản phẩm kháng sinh của Công ty TNHH Đổi mới Dược phẩm, đang trong một thử nghiệm lâm sàng giai đoạn II.
- Kevzara: Sanofi và Regeneron đã bắt đầu thử nghiệm an toàn giai đoạn I để kiểm tra xem Kevzara, một loại thuốc chống viêm được phê duyệt, có thể làm giảm các triệu chứng của COVID-19 hay không.
Nghiên cứu thuốc tiền lâm sàng COVID-19
Thuật ngữ "nghiên cứu tiền lâm sàng" được định nghĩa bởi các nghiên cứu trong phòng thí nghiệm in vitro và in vivo, chỉ ra giai đoạn đầu phát triển vắc-xin, kháng vi-rút hoặc kháng thể đơn dòng, như thí nghiệm để xác định liều lượng và độc tính hiệu quả, trước một hợp chất ứng cử viên được nâng cao để đánh giá sự an toàn và hiệu quả ở người. Để hoàn thành giai đoạn phát triển thuốc tiền lâm sàng – sau đó được kiểm tra về tính an toàn và hiệu quả ở một số người bị nhiễm COVID-19 (hàng trăm đến hàng ngàn ở các quốc gia khác nhau) – là một quá trình có thể cần 1-2 năm cho COVID-19 vắc-xin và liệu pháp, theo một số báo cáo vào đầu năm 2020. Bất chấp những nỗ lực này, tỷ lệ thành công để các ứng cử viên thuốc đạt được sự chấp thuận theo quy định cuối cùng thông qua quy trình phát triển thuốc để điều trị các bệnh truyền nhiễm là 19% và đối với các ứng cử viên vắc-xin cụ thể, chỉ có 11,5%.
Các chất ức chế

Vào tháng 3 năm 2020, protease chính của virus SARS-CoV-2 được xác định là mục tiêu của các loại thuốc sau nhiễm trùng. Enzyme này rất cần thiết cho tế bào chủ để tái tạo axit ribonucleic của virus. Để tìm ra enzyme này, các nhà khoa học đã sử dụng bộ gen được các nhà nghiên cứu Trung Quốc công bố vào tháng 1 năm 2020 để phân lập protease chính.
Ứng cử viên kháng vi-rút, miễn dịch và kháng thể
Vào tháng 3 năm 2020, IFN-alpha và umifenovir đã được phát triển trong nghiên cứu giai đoạn đầu với tư cách là thuốc chống vi rút sau nhiễm trùng. Nhiều ứng cử viên khác, bao gồm liệu pháp miễn dịch và các hợp chất kháng thể, đang được phát triển.